Ion semiconductor sequencing technology is used for Ion Torrent (Thermo Fisher) sequencers.
Sequencing-by-synthesis method and emulsion PCR (emPCR) is used but no light (no optics; post-light sequencing) is used. Instead of fluorescence or chemiluminescence, H+ ions released during base incorporation is measured.
It offers sequencing within 2-3 hours, rapid turnaround time (~2 days) from sample to DNA sequences, high Accuracy: 99.97%.
(Ion PGM sequencer)
(Ion S5 sequencer))
The steps occurring in the sequencer include primer annealing, polymerase binding, and chip loading.
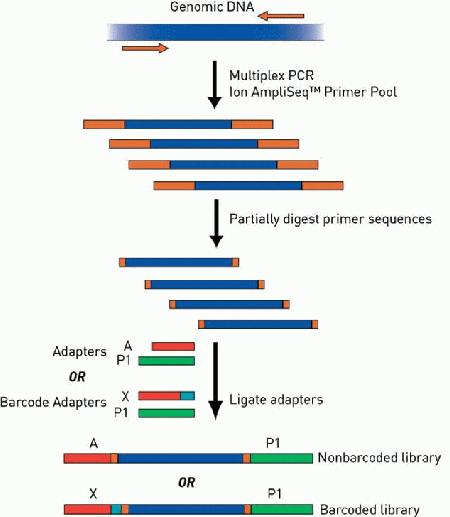
Kits: library kit, template kit, sequencing kit. The library kit is based on 200bp chemistry and includes barcodes to enable the cost-effective and flexible processing of samples. The barcode adapters contain sequences, and they enable pooling up to 24 amplicon libraries and to conduct multiplexed NGS analysis, reducing the sequencing cost per sample.
Life Technologies/ Thermo Fisher NGS portfolio :
Ion Personal Genome Machine (PGM) sequencer---device cost $50k , run cost $300 to $750, output capability 1Gb, speed of runs 2 hours, read length 200b. For this sequencer, 10 ng input DNA needed. Consensus accuracy 99.99%
Ion Proton™-----device $149k, first chip PI, is able to generate ~10Gb per run while the PII is able to generate ~100Gb per run. Its for whole genome sequencing.
S5 system is superior to Proton or the PGM. S5 deals with Indel issues better. With the Ion Torrent Chef's library construction functionality, eight AmpliSeq libraries can be prepared in an eight-hour run. Combined with Ion Torrent AmpliSeq technology for target selection, the Ion Chef System for automated library and template preparation, and Ion Reporter Software for automated variant annotation, targeted sequencing is easy.
Workflow consists of four major steps: library construction (construct library), template prep (prepare template), sequencing (run sequence) and analysis (analyze data).
Library Construction: taking DNA (or RNA converted to DNA), fragmenting it to a uniform size (generally 200-400b), adding sequencing adapters.
Template Prep/Amplification: fragments generated during the library prep are attached to beads and amplified using emulsion PCR. Beads coated with complementary primers are mixed with a dilute aqueous solution containing the fragments to be sequenced along with the necessary PCR reagents. This solution is then mixed with oil to form an emulsion of microdroplets. The concentration of beads and fragments is kept low enough such that each microdroplet contains only one of each. Clonal amplification of each fragment is then performed within the microdroplets. Following amplification the emulsion is ‘broken’ and the amplified beads are enriched in a glycerol gradient.
(http://www.thermofisher.com/us/en/home/technical-resources/research-tools/image-gallery/image-gallery-detail.19800.html)
Sequencing: Individual bases are introduced one at a time and incorporated by DNA polymerase. The direct release of H+ (protons) from the reaction is measured. The chips act as pH meters and are disposable. Image scans are not needed, so sequencing reactions are relatively fast, with 200b reads taking about 2 hours.
#Error rates are ~1%, but pyrosequencing chemistry has trouble with long homopolymers (e.g., AAAAAA). Stretches of the same base will result in a single, but stronger, signal.
Data analysis: Standard output files are FASTQ, BAM, SFF, VCF. Torrent Browser’ software is main interface, while cloud-based solution ‘Ion Reporter’ is used for data analysis.
Chip:
The chip has a dense array of >1 million micro-machined wells with ion sensor. Each well contains a different DNA template, allowing massively parallel sequencing. 1 chip is used per run. Beneath the wells is an ion-sensitive layer and a proprietary Ion sensor at the bottom.
Three chips are available for the S5 instruments: the 520, 530, and 540, all of which have 2.5-hour sequencing run times for 200 base single-end reads.
The 520 chip generates between 1Gb to 2 Gb of data, or 3 million to 5 million reads with read lengths up to 400 bases. Analysis takes five hours on the S5 and one-and-a-half hours on the XL. The 520 chip is geared primarily toward gene panels.
The 530 chip generates between 3 Gb and 5 Gb of data, or 15 million to 20 million reads with read lengths up to 400 bases. The standard eight-hour analysis time can be shortened to two-and-a-half hours with the XL.
The 540 chip generates between 10 Gb to 15 Gb of data, or 60 million to 80 million reads. It does not yet support 400-base reads. Analysis time is 16.5 hours on the S5 and five hours with the XL. The 540 chip is similar to the PI chip on the Proton, able to sequence exomes and transcriptomes, as well as larger gene panels.
The microprocessor chips generate fewer reads than HiSeq or SOLiD, so it's used was confined to small genomes and targeted sequencing.
(http://www.genomics.cn/en/navigation/show_navigation?nid=2640)
(https://www.researchgate.net/publication/255715498_Single-Cell_Semiconductor_Sequencing)
(http://www.genomics.cn/en/navigation/show_navigation?nid=2640)
(http://allseq.com/knowledge-bank/sequencing-platforms/ion-torrent/)
#If problem occurs, shut down the system and server and reboot them. The server may enter a system check after the reboot, which will take 3-4 hours. If you have a monitor and keyboard connected directly to the Torrent Server, you can avoid the system check by pressing “c” on the keyboard. The data will automatically transfer to the server after connection.
Uses of this sequencer include, exome sequencing, noninvasive prenatal testing, variant detection by targeted gene sequencing in cancer and genetic disorders, characterizing novel microbes, typing of pathogens.